DNAやmRNAのエタ沈やイソプロ沈で収率が悪くなって困っていませんか?回収率を上げてロスしないコツはこちら

SYBR GREEN法によるqPCRのプライマー設計に、PRIMER3などの専用ソフトを利用しているラボも多いと思います。
ですが、そのような専用ソフトやフリーソフトを使用しなくても、WEB上でプライマーを設計できてしまう方法をご紹介したいと思います。
習うより慣れろです。
まずは試してみましょう。
PubMedで知られているサイトです⇒https://www.ncbi.nlm.nih.gov/
検索窓の左の選択肢を「Nucleotide」にして、遺伝子名(タンパク質名でもOK)を入力
ここでは例として「Cidea」を検索してみましょう。
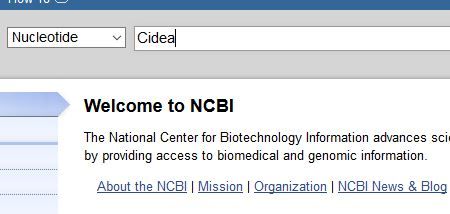
左の「Molecule types」内の「mRNA」をクリック。
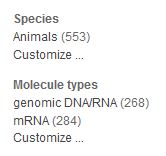
右の「Top Organisms」で動物種(Mus musclulus)をクリック。
知りたい遺伝子をはじめ、関連遺伝子などの結果一覧が返ってきます。
それぞれに、Accession numberがついています。
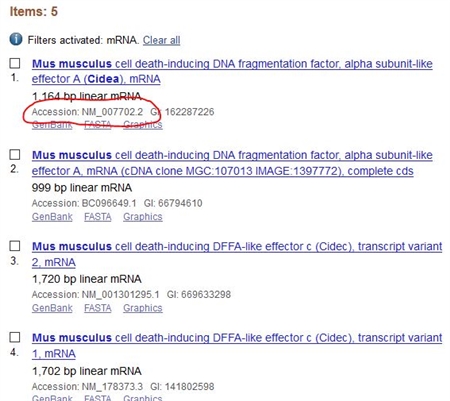
マウスのCidea mRNAを検索したら、このような結果になりました。
mRNAと完全cdsが登録されていますね。
ここでは、mRNAを選んでみましょう。
遺伝子名をクリックしたら、下のように、その遺伝子の詳細のページに移ります。
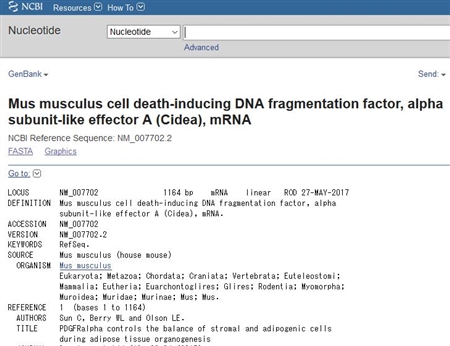
下のほうにスクロールしていくと、その遺伝子の機能が参考論文とともに記述されています。
また、このmRNAの全シークエンスも書かれています。
また、右メニューにも、様々な情報へのリンクがあります。
これらも重要な情報ですが、プライマーの設計では使いません。
ここで、右メニューにある「Pick Primers」をクリックします。
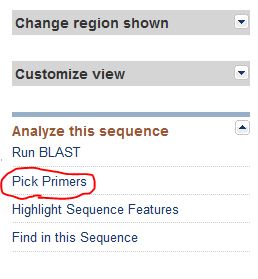
プライマー設計の条件を具体的に入力する画面に移ります。
一番上の入力欄に、すでにCideaのアクセッションナンバーが反映されています。
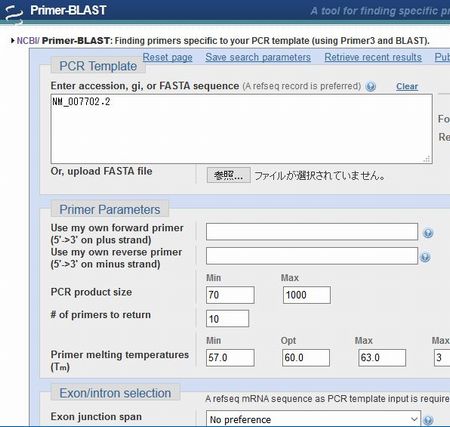
それでは、プライマー検索の各条件を入力していきましょう。
以下は私がSYBR GREEN法のqPCRで主に使う検索条件です(一つの参考にしてください)。
なお、以下に記載のない部分の条件はデフォルトの条件のままです。
※「Exon〜」と「Intron inclusion」は、どちらから一方だけで構いません。どちらもゲノムDNAの増幅を避けるための(もしくはゲノムDNAからの産物と混同しないための)選択です。
さらに詳細条件を入力
Advanced parametersをクリックします。
最後に、「Use new graphic view」にチェックを入れ、「Get Primers」をクリックして検索開始です。
しばらく待っていると、候補となるプライマーが複数提示されます。
ポイントは、あまり条件を厳しくし過ぎないことです。
あまり厳しくすると、結果が返って来ないことが多いです。
条件は多少緩めに設定し、出てきた結果を自分でチェックし、最適なものを選ぶようにしたほうが良いと思います。
ちなみに、下記のサイトがプライマーの条件設定の決定に役立ちます。
http://www.takara-bio.co.jp/prt/pdfs/prt3-1.pdf
http://www.takara-bio.co.jp/prt/pdf/prt1-2.pdf
こんな感じでプライマー候補の結果が表示されます。
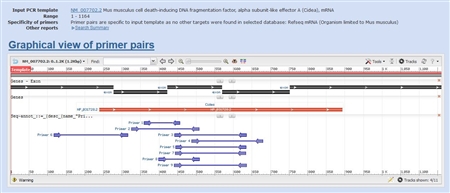
上段の黒いバーが各エクソンを表しています。
中段の赤いバーがアミノ酸に翻訳させる領域です。
下段の矢印とそれに挟まれた領域が、プライマーがアニーリングする位置と、増幅PCR産物を表しています。
上記の例ですと、どのPCR産物もエクソンとエクソンのつなぎ目を含んでいることに着目してください。
下にスクロールしていくと、プライマー候補の配列が表示されます。

プライマー配列の情報が重要なのはもちろんですが・・・
たくさんある候補のうち、どれを選べば良いか、迷ってしまいますよね?
ということで、判別するポイントをお伝えします。
ポイント1:ミスマッチの存在する配列は避ける
通常、結果には次のように塩基配列が100%マッチしているプライマーが表示されます。

100%マッチしている場合は、「Template」が「・・・・・・・」となっています。
ですが、時々、100%マッチに加えて、塩基配列が少しだけ合っていない候補も表示される場合があります。
(合っていない配列は「・」が正しい塩基になっています)
このようなミスマッチ配列が検出されるプライマー候補は避けるべきです。
ポイント2:プライマーの自己結合が生じそうなものを避ける
「Self Complementarity」「Self 3' complementarity」に着目してください。

プライマー内部や、プライマー同士で相補的な塩基配列が多いと、プライマ―自身で2次構造を形成したり、プライマーのダイマーができてしまい、PCR反応がうまく行われない場合があります。
上記の数値は、そのプライマー内部や3末での相補配列の数を表しています。
少ないほどベターです。
一応、
Self Complementarityは2以下
Self 3' complementarityは1以下
が理想ですが、実際にはなかなかそんな配列はヒットしません。
また、これらの数値が大きくても、問題なく使えるプライマーもたくさん経験しています。
ですので、プライマーがワークするかどうか、やってみなければ分からない、という面も多いのですが、
数値が少ないものを優先的に選ぶようにします。
なお、FとRのプライマー間の相補性は表示されていません。
ポイント3:3'末端がT以外のものを選ぶ
Tだと、ミスマッチ配列でも反応してしまう可能性が高いようです。
ポイント4:FとRプライマーのTmの差が少ないものを選ぶ
もっとも、1℃未満に設定しているので、あまり影響はないかもしれませんが・・・
私は次のところをゆるめに設定することが多いです。
実際のところ、プライマーは使ってみて初めて良し悪しが分かります。
何本かに1本は外れの配列あっても仕方ないです(上の条件を使えばそんなに外れませんが)。
というか、何回かに1回は外れるものだ、そういうものだ、外れたらもう一度設計すればいいだけ、と考えると精神衛生的には良いです。
実際、プライマーは安価ですし、発注すればすぐに届きますし。
まず、qPCRが終わった後の融解曲線をチェックします。
理想は大きなピークが1つあることです。
低温側(左側)に中程度のピークが見られる場合がありますが、これはプライマー同士の結合による増幅産物の可能性が高いです。これは失敗です。
また、融解曲線の大きなピークが2つある場合、片方はイントロンを含んだPCR産物、つまりmRNAではなくゲノムDNAの増幅産物が含まれている可能性があります。
qPCRを実施後のアンプリコン(PCR産物)をDNAラダーとともに1%アガロースゲルで電気泳動を行い、出るはずの位置にバンドが1つだけ出ていることを確認します。
予想される位置以外の場所にバンドが出る場合、qPCRの結果はノンスペのPCR産物の結果であるので使えません。
かなり低い位置にバンドが見られる場合はプライマー自身の増幅産物、高い位置にある場合は、エクソンを含むPCRと考えられ、ゲノムDNAが反応していると思われます。
全長mRNAの場合は3'末端、5'末端のUTR(非翻訳領域)も含む、mRNA全体の塩基配列ですが、CDSの場合はUTRを含んでいないコーディング領域のみです。
プライマー設計する場合は、どちらを使っても理論上問題となることはないと思います。
私個人的には、mRNAを好んで使っています。
cDNA合成に使われるdTプライマーは、mRNAの3'末端にあるポリA領域から逆転写を開始します。
このことから、出来上がるcDNAは、3'末端寄りのものがメインになります。
qPCR用プライマーをあまり5'末端寄りに設計すると、検出効率が悪くなってしまいます。
だいたいの目安として、3'末端から1.5kb以内のところに設計するようにしましょう。
なお、逆転写にランダムプライマーを使っている場合はこの問題はありません。
使っているcDNA合成キットをチェックしてみてくださいね!